Images
Científicos de Estados Unidos, Canadá y España, entre ellos Joan Guinovart del IRB Barcelona, investigarán unidos para hallar un tratamiento para la enfermedad de Lafora, la epilepsia más severa en humanos.
El Instituto de Salud de los Estados Unidos otorga al consorcio 8,5 millones de dólares durante 5 años.
La asociación de familias afectadas Chelsea’s Hope de Estados Unidos ha propiciado la colaboración internacional.
El laboratorio de Joan J. Guinovart en el Instituto de Investigación Biomédica (IRB Barcelona) forma parte del consorcio mundial que acaba de iniciar su andadura para hallar un tratamiento contra la enfermedad de Lafora, la forma más severa de epilepsia en humanos.
El proyecto liderado por la Universidad de Kentucky, en Estados Unidos, ha recibido financiación de los Institutos de Salud de los Estados Unidos (National Institutes of Health –NIH-) por 8,5 millones de dólares (7,7 M€) para los próximos cinco años. La gestación del proyecto la propició Chelsea’s Hope, asociación de familias afectadas por Lafora en Estados Unidos, que en 2014 reunió a los principales especialistas que estudian la enfermedad desde distintos ángulos.
La enfermedad de Lafora es una patología neurodegenerativa hereditaria que se manifiesta con la primera crisis epiléptica en la adolescencia, entre los 10 y los 17 años, y no tiene tratamiento. Su evolución está marcada por una degeneración progresiva del sistema nervioso que sume al paciente en un estado vegetativo, causando la muerte unos diez años después de su aparición.
El equipo de investigación está integrado por los laboratorios de Matthew Gentry, de la Universidad de Kentucky –líder del consorcio- con quien colaborará Pascual Sanz del Instituto de Biomedicina de Valencia del CSIC, Berge Minassian del Hospital for Sick Kids en Toronto, Canadá, Peter Roach de la Universidad de Indiana, en Estados Unidos, José Serratosa del Hospital Universitario Fundación Jiménez Díaz de Madrid y Joan Guinovart del IRB Barcelona y catedrático de la Universidad de Barcelona.
“Todos llevamos trabajando más de 10 años en Lafora y ahora lo haremos juntos para tratar de lograr algo muy importante. Es maravilloso que el empuje haya salido de las familias”, destaca Joan Guinovart. “Solo uniendo fuerzas podremos encontrar soluciones para enfermedades raras como la que nos ocupa”, opina.
¿Qué sabemos de Lafora?
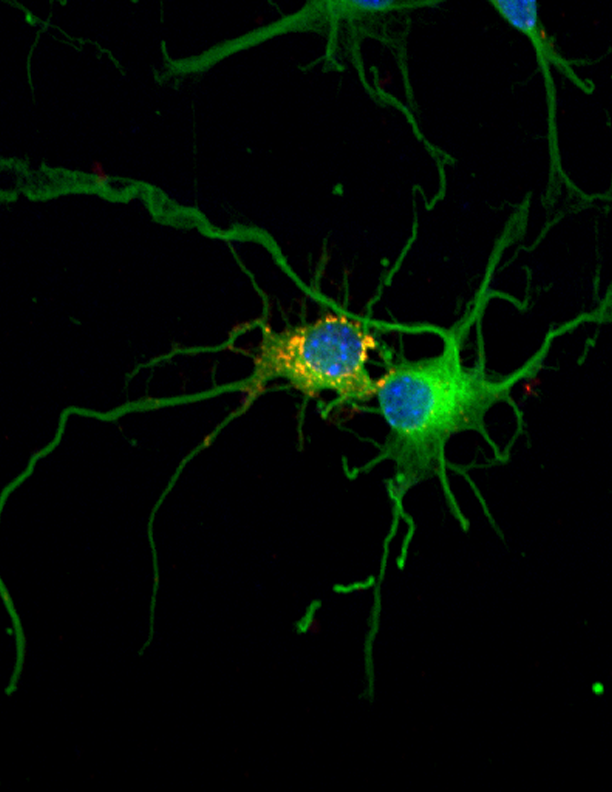
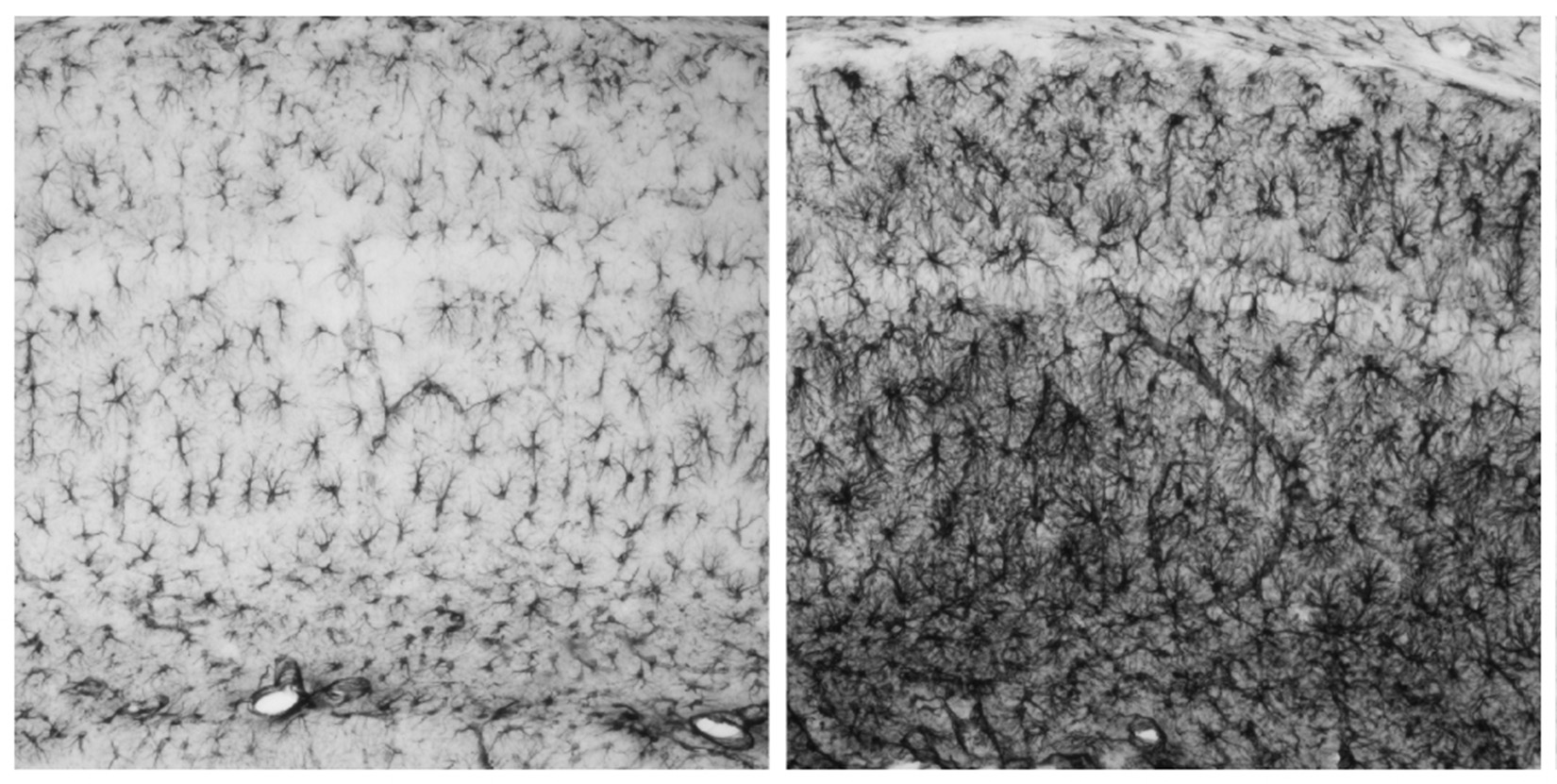
“Es la primera epilepsia de la que se conocen las bases moleculares”, explica Guinovart. Se transmite de forma hereditaria a través de padres portadores de mutaciones en uno de los dos genes relacionados con la enfermedad. Estos genes son la laforina (nombrada así en honor al Dr. Lafora, neurólogo español descubridor de la enfermedad) y la malina (del término francés le grand mal utilizado para referirse a epilepsias). La enfermedad se caracteriza por la acumulación en las neuronas de unos depósitos anormales llamados “cuerpos de Lafora”.
Guinovart y su equipo, especialistas en el metabolismo del glucógeno, demostraron en 2007 en un artículo en Nature Neuroscience que mutaciones en cualquiera de estos dos genes provocan la acumulación de glucógeno –cadenas de azúcar- en las neuronas, lo que les es tóxico y precipita su muerte celular. Asimismo, demostraron en ratones que bloqueando la síntesis de glucógeno en las neuronas, la enfermedad no se desencadena, identificando así un posible tratamiento para Lafora.
“Ahora tenemos que ser capaces de ver si una vez manifestada la enfermedad y bloqueando la síntesis de glucógeno podemos frenar su progresión o incluso revertirla. También exploraremos vías para prevenirla”, describe.
Cada laboratorio se focalizará en aspectos diferentes de la enfermedad, desde la ciencia básica hasta la ciencia traslacional, con el objetivo de que sus descubrimientos puedan traducirse en el diseño de un ensayo clínico con pacientes.
What do we know about Lafora disease?
“It is the first type of epilepsy for which the molecular bases are known,” explains Guinovart. It is transmitted when both parents pass on a mutation in one of the two genes involved with the disease, namely laforin (named after Dr. Lafora, a Spanish neurologist who discovered the disease) and malin (from the French term le grand mal used to refer to epilepsies). The disease is characterised by the accumulation of abnormal deposits, called Lafora bodies, in neurons.”
In an article published in Nature Neuroscience in 2007, Guinovart and the members of his team, experts in glycogen metabolism, demonstrated that mutations in either of these two genes lead to the accumulation of glycogen—chains of sugar—in neurons, thus causing toxicity and triggering neuronal death. Likewise, using mice, they showed that the inhibition of glycogen synthesis in neurons prevents the development of Lafora disease, thereby identifying a possible treatment for this condition.
“We now have to study whether blocking glycogen synthesis after the onset of the disease can halt its progression or even reverse the condition. We are also looking into ways to prevent the disease,” he says.
Each lab will focus on different aspects of the disease, covering from basic to translational research, with the aim to make breakthroughs that can be used to design a clinical assay involving patients.
IRB Barcelona
El Instituto de Investigación Biomédica (IRB Barcelona) trabaja para conseguir una vida libre de enfermedades. Desarrolla una investigación multidisciplinar de excelencia para curar el cáncer y otras enfermedades vinculadas al envejecimiento. Establece colaboraciones con la industria farmacéutica y los principales hospitales para hacer llegar los resultados de la investigación a la sociedad, a través de la transferencia de tecnología, y realiza diferentes iniciativas de divulgación científica para mantener un diálogo abierto con la ciudadanía. El IRB Barcelona es un centro internacional que acoge alrededor de 400 científicos de más de 30 nacionalidades. Reconocido como Centro de Excelencia Severo Ochoa desde 2011, es un centro CERCA y miembro del Barcelona Institute of Science and Technology (BIST).





