
Professor (Biochemistry and Molecular Biology Dept. - UB), CIBERDEM ISCIII
Research information
Background
Age-related metabolic diseases have increased to epidemic proportions in all industrialized countries. Aging is the most universal contributor to the aetiologies of metabolic decline and related diseases, including type 2 diabetes mellitus, cardiovascular disease, non-alcoholic steatohepatitis (NASH), stroke, neurodegenerative diseases, and cancer. According to observational studies, obesity is a major determinant of premature mortality and a risk factor for leading causes of death. Diabetes has increased dramatically over the past century, registering of 371 million cases worldwide in 2012. The prevalence of diabetes mellitus and glucose intolerance increases significantly with age.
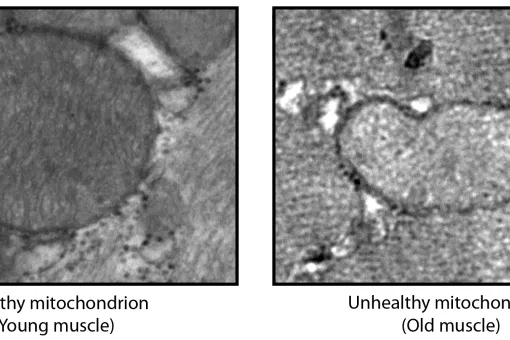
Mitochondrial dysfunction has been reported in complex age-related diseases, as well as in aging itself. These observations thus support the notion that mitochondrial dysfunction and the accumulation of oxidative damage contribute to the pathogenesis of these diseases. Aging is characterised by the accumulation of mutations in mitochondrial DNA, which may be responsible for the alterations detected in this condition. In addition, the deregulation of various signalling pathways can induce mitochondrial dysfunction. The regulatory network that governs mitochondrial homeostasis may therefore be involved in the development of age-related conditions.
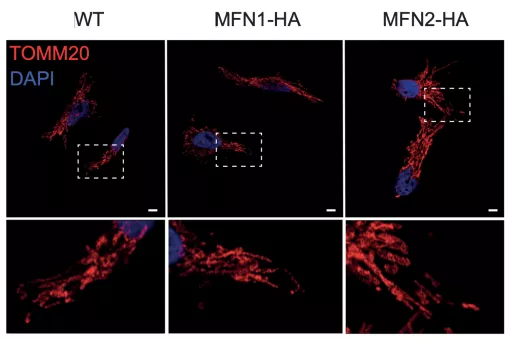
We have shown that mitochondrial morphology determines the function of these organelles. Mitochondrial architecture is determined by motility, fusion, and fission events. Mitochondrial fusion is mediated by mitofusins (Mfn1 and Mfn2, located in the outer mitochondrial membrane) and optic atrophy gene 1 (Opa1, located in the inner membrane), while mitochondrial fission is mediated by dynamin-related protein 1 (Drp1), mitochondrial fission factor (Mff, located in the outer mitochondrial membrane), and fission 1 protein (Fis1, located in the outer mitochondrial membrane), among others.
Some of the proteins involved in mitochondrial dynamics are subject to exquisite control. In this regard, we have reported that the expression of Mfn2 is highly regulated in response to enhanced energy expenditure and exercise. Mfn2 is repressed in the muscle of obese or type 2 diabetic patients. The nuclear co-activators PGC-1alpha, and PGC-1beta activate Mfn2 gene transcription through ERRalpha and may participate in the changes in the expression of this gene. OPA1 and mitochondrial elongation are enhanced by insulin in cardiomyocytes.
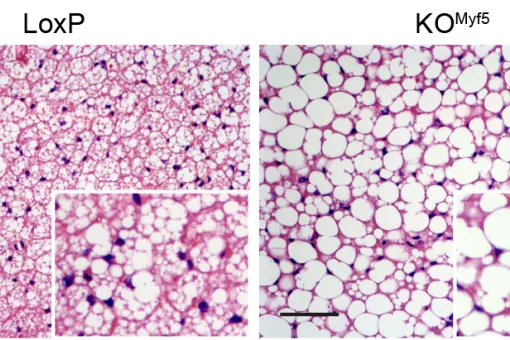
In addition, we have reported that some proteins involved in mitochondrial fusion modulate metabolism in vivo. Mnf2 deficiency enhances proton leakage and reduces respiration, independently of changes in mitochondrial mass. Mfn2 ablation also alters glucose metabolism and energy expenditure. Liver-specific ablation of Mfn2 in mice leads to glucose intolerance and enhanced hepatic gluconeogenesis, whereas ablation in muscle causes higher susceptibility to glucose intolerance and insulin resistance in response to a high fat diet. Hypothalamic POMC neuron-specific ablation of Mfn2 results in hyperphagia, reduced energy expenditure, and obesity. OMA1 (an Opa1 protease) knockout mice are also obese as a result of deficient brown adipose tissue metabolism.
As to the mechanisms controlled by Mfn2, this protein regulates endoplasmic reticulum (ER) morphology, and the transfer of calcium from the ER to the mitochondria. We have also demonstrated that Mfn2 deficiency causes a chronic Unfolded Protein Response (UPR), which is crucial in the metabolic alterations associated with this condition.
Mitochondrial dynamics also plays a key regulatory role in determining the fate of mitochondria, namely whether they undergo degradation via selective autophagy.
Research interests
We aim to identify the mechanisms by which mitochondrial dysfunction participates in the development of complex metabolic disorders such as obesity, insulin resistance, and type 2 diabetes. More specifically, we focus on the implications of mitochondrial fusion or mitochondrial fission proteins in age-related diseases, and the role of interplay between autophagy, mitochondrial function, and energy metabolism.
Research lines
Our group is organised around 3 research lines:
1. Analysis of the implications of proteins involved in mitochondrial dynamics on the development of age-related diseases.
We have demonstrated that proteins participating in mitochondrial fusion, such as Mfn2 and the protease OMA1, are relevant regulators of metabolism and that their deficiency causes alterations such as insulin resistance or obesity. Thus, we have reported that Mfn2 in liver, muscle, and the hypothalamus plays a key role in metabolic homeostasis. Mfn2 deficiency represses mitochondrial respiration and triggers ROS production and endoplasmic reticulum stress in various cell contexts. These effects lead to deficient insulin signalling in skeletal muscle and liver, and to glucose intolerance.
We are currently using cell-based models and knockout mice to study the metabolic role of other relevant proteins in mitochondrial dynamics. In addition, we are analysing the mechanisms by which mitochondrial dynamics proteins alter mitochondrial function, paying special attention to the function of the protein on the mitochondrial-endoplasmic reticulum connections.
2. Role of interplay between autophagy, mitochondrial function, and energy metabolism.
We have identified a new regulator of autophagy named DOR (also TP53INP2), which operates in mammalian cells, flies, and mouse skeletal muscle. Found in autophagosomes under conditions of activated autophagy, DOR binds to Atg8 proteins, LC3, and GATE16. In addition, it activates autophagy in mammalian cells and in mouse skeletal muscle. Data from our laboratory also indicate that DOR regulates muscle mass in mice via the modulation of autophagy.
At present, we are addressing whether DOR participates in energy metabolism and in normal mitochondrial function. IN this regard, we plan to perform a specific analysis of the molecular mechanisms of DOR functions.
3. Identification of novel targets and development of new compounds for the treatment of metabolic disorders.
We aim to explore the innovation capacities of our mechanistic findings. At present, our therapeutic hypothesis is that the activation of Mfn2 will produce effects in tissues, which will be of therapeutic benefit to diabetic or obese patients and those affected by Charcot-Marie-Tooth type 2A neuropathy (caused by mutations in the MFN2 gene).
We have developed and validated a screening strategy to search for activators of Mfn2 in human cells. This screening protocol with a small chemical library has allowed the identification of a compound that induces Mfn2 expression. Studies in human and in mouse cell cultures have demonstrated that this compound increases the elongation of mitochondria, enhances the expression of Mfn2, and boostsmitochondrial membrane potential. These observations provide a proof of concept for the future identification of Mfn2 activators through our screening method.
Selected publications
Projects
“Novel gene-therapy for the treatment of AATD and liver-related diseases”, financiado por la Agència d’Ajuts Universitaris i de Recerca mediante la convocatoria d’Ajuts d’Indústria del Coneixement per a l’any 2024. Referencia: 2024 LLAV 00107


"Implicaciones de la dinámica mitocondrial y la autofagia sobre el metabolismo energético" financiado por el Ministerio de Ciencia e Innovación- Agencia Estatal de Investigación y por el Fondo Europeo de Desarrollo Regional (FEDER) de la Unión Europea. Referencia: SAF2016-75246-R.
“Producción industrial en células de omega-3 (dha y epa) para su uso en alimentación”, financiado por el Ministerio de Ciencia, Innovación y Universidades- Agencia Estatal de Investigación. Retos – Colaboración. RTC2019-007154-2.
"Phospholipid biosynthesis and transport between endoplasmic reticulum and mitochondria: understanding their essential role in liver disorders", funded by La Caixa Foundation. Reference: HR21-00430.

"Desarrollo de fármacos primeros en su clase para tratar la esteatohepatitis no alcohólica (Drug4NASH)", PDC2021-121010-I00, funded by MCIN/AEI/10.13039/501100011033 and by the European Union “NextGenerationEU”/PRTR”
"Papel de proteínas de fusión mitocondrial y de la autofagia en el control del metabolismo y la inflamación, e impacto en patología" PID2019-106209RB-I00, financiado por MCIN/ AEI/10.13039/501100011033.
"Lipodystrophy and sarcopenia in Hutchinson-Gilford progeria syndrome: Mechanisms and role in disease progression" PID2019-106209RB-I00, financiado por La Marató de TV3. Referencia: 202033-30-31.

"Drug4NASH: First-in-Class drugs to treat non-alcoholic steatohepatitis (NASH)", finançat per l’Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR) a través de la convocatòria dels ajuts d'Indústria del Coneixement per a l'any 2021 (Llavor i Producte). Referència: 2021 PROD 00045.

"Metabolic diseases: Structural and functional features of membrane proteins and inter-organelle contacts" Grup de Recerca consolidat (SGR-Cat 2021) del Departament de Recerca i Universitats. Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR). Referència: 2021 SGR 01281.
"Identificación de moduladores del metabolismo mitocondrial e impacto en patología" financiado por MCIN/AEI/10.13039/501100011033/FEDER, UE a través de la convocatoria «Proyectos de Generación de Conocimiento 2022». Referencia: PID2022-137576OB-I00.