
Professor (Organic Chemistry Dept. - UB)
Meet Our Scientists Videos
Research information
Background
Most of the compounds associated with living organisms, such as proteins, carbohydrates or nucleic acids are chiral. Chiral molecules are those that are not superimposable on their mirror image. Consequently, there can be two distinct isomers of these molecules, called enantiomers. Enantiomers of compounds usually have different biological activity due to the different interaction with chiral biomolecules. Since a drug must match the receptor in the cell, it is often only one of the enantiomers that is of interest. In certain cases the other enantiomer may be harmful. Consequently, nowadays most drugs are either achiral or enantiomerically pure.
Asymmetric synthesis is a chemical methodology that produces only one enantiomer of the chiral compound with high selectivity using chiral reagents or catalysts. In a catalytic asymmetric reaction, a small amount of chiral catalyst is used to produce large quantities of an enantiomerically enriched compound from an achiral precursor. Such process can be highly efficient and environmentally clean. Industrial companies are concerned about disposing of unwanted compounds and also about the inefficiency and costs involved in the chemical processes. As a result, there is a strong demand for asymmetric syntheses methods.
The research of the group is devoted to the synthesis of biologically active compounds. In the case of chiral molecules, asymmetric catalysis is, arguably, the most efficient method. Therefore, the group also work in the development of new catalytic asymmetric methodologies.
Research interests
The general objective of the group is the preparation of compounds of biological or pharmacological interest. To this end we are working in two main areas: the development of new synthetic methods with the focus in asymmetric catalysis and the synthesis of new compounds designed for a biological function.
Research lines
1. Development of new synthetic methods.
- 1.1 Research on the Pauson-Khand reaction (PKR). This is one of our oldest research lines and dates back to 1992. The Pauson-Khand reaction is a cobalt-catalyzed cycloaddition between an alkyne, an alkene and carbon monoxide to give a cyclopentenone (Figure 1).
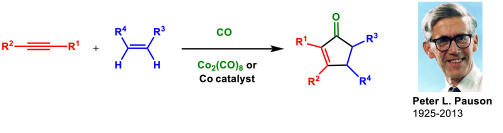 Figure 1. The Pauson-Khand reaction.
Figure 1. The Pauson-Khand reaction.
We have made many contributions to this subject (69 articles). Among them we could highlight the development the first cobalt-catalyzed asymmetric version of an intermolecular PKR using a chiral diphosphane (ThaxPHOS) designed in our group for such purpose (Figure 2).
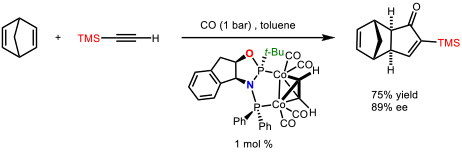 Figure 2. First catalytic asymmetric version of an intermolecular Pauson-Khand reaction. (Orgue, S.; Leon, T.; Riera, A.; Verdaguer, X. Org. Lett. 2015, 17, 250-253)
Figure 2. First catalytic asymmetric version of an intermolecular Pauson-Khand reaction. (Orgue, S.; Leon, T.; Riera, A.; Verdaguer, X. Org. Lett. 2015, 17, 250-253)
- 1.2 Development of new catalytic methodologies. In the last years, we focussed on isomerization reactions of strained heterocycles since they show perfect atom economy. The iridium-catalyzed, isomerization of N-sulfonyl aziridines, using the well know Crabtree’s catalyst, allowed the preparation of allyl amines in a straightforward manner (Figure 3).
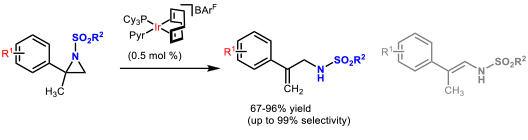 Figure 3. Iridium-catalyzed isomerization of N-sulfonyl aziridines to allyl amines. (Cabre, A.; Sciortino, G.; Ujaque, G.; Verdaguer, X.; Lledos, A.; Riera, A. Org. Lett. 2018, 20, 5747-5751)
Figure 3. Iridium-catalyzed isomerization of N-sulfonyl aziridines to allyl amines. (Cabre, A.; Sciortino, G.; Ujaque, G.; Verdaguer, X.; Lledos, A.; Riera, A. Org. Lett. 2018, 20, 5747-5751)
One of the most innovative catalytic reactions developed by our group is the regioselective isomerization of 2,2-disubstituted oxetanes to homoallylic alcohols. which were enantioselectively hydrogenated to chiral 2-aryl-2-methylpropanols (Figure 4).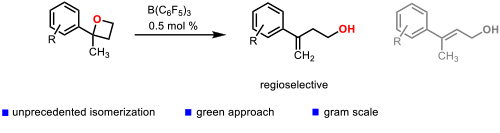 Figure 4. Catalytic isomerization of 2,2-disubstituted oxetanes to homoallylic alcohols. (Cabre, A.; Rafael, S.; Sciortino, G.; Ujaque, G.; Verdaguer, X.; Lledos, A.; Riera, A. Angew. Chem., Int. Ed. 2020, 59, 7521-7527).
Figure 4. Catalytic isomerization of 2,2-disubstituted oxetanes to homoallylic alcohols. (Cabre, A.; Rafael, S.; Sciortino, G.; Ujaque, G.; Verdaguer, X.; Lledos, A.; Riera, A. Angew. Chem., Int. Ed. 2020, 59, 7521-7527).
- 1.3 New chiral ligands for to asymmetric catalysis. Since most marketed drugs are chiral and are prescribed as a single enantiomer, the development of new methods of asymmetric catalysis is of great importance for the chemical and pharmaceutical industry. In the development of synthetic methodology, our main focus has been on enantioselective reactions catalyzed by organometallic complexes. Such catalysts induce chirality by means of chiral ligands bound to the metal. Our main area of research is the development of new chiral ligands that can coordinate to metals and their application to asymmetric catalysis. We are renowned experts in the synthesis of P-stereogenic phosphines. We have developed stereoselective methodologies for the synthesis of the tert-butylmethylphosphine fragment and its coupling with amines (Figure 5).
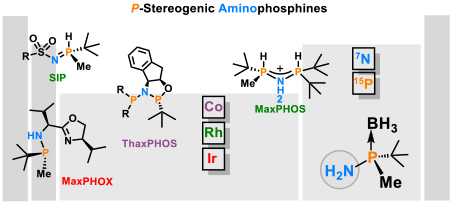 Figure 5. Chiral ligands developed in the group. (Cabre, A.; Riera, A.; Verdaguer, X. Acc. Chem. Res. 2020, 53, 676-689).
Figure 5. Chiral ligands developed in the group. (Cabre, A.; Riera, A.; Verdaguer, X. Acc. Chem. Res. 2020, 53, 676-689).
Our greatest recent accomplishment has been the development of a family of ligands that we called MaxPHOX (Figure 6). The synthesis was based on the methodology previously developed in the group.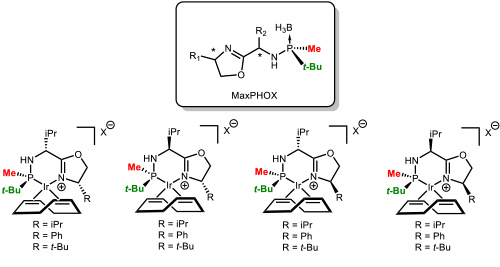 Figure 6. The family of MaxPHOX ligands. (Salomo, E.; Orgue, S.; Riera, A.; Verdaguer, X. Angew. Chem., Int. Ed. 2016, 55, 7988-7992).
Figure 6. The family of MaxPHOX ligands. (Salomo, E.; Orgue, S.; Riera, A.; Verdaguer, X. Angew. Chem., Int. Ed. 2016, 55, 7988-7992).
- 1.4 Asymmetric hydrogenations and isomerizations. The iridium complexes of MaxPHOX ligands proved to be excellent catalysts for isomerization or hydrogenation reactions. Moreover, as MaxPHOX are a library of ligands, we were able to select the best catalyst for each case, with the subsequent optimization of their performance. Somewhat surprisingly, we found that each reaction required a different catalyst of the family. As catalysts for asymmetric hydrogenation, the performance of the iridium complexes of MaxPHOX has been extraordinary, showing good to excellent enantiomeric excesses in the asymmetric hydrogenation of N-sulfonyl allyl amines (1) to give β-methyl amines, as well as in the asymmetric hydrogenation of N-aryl imines (2, R2 = Ar). It is worth noting that, in the asymmetric hydrogenation of cyclic enamides (3), 2-aryl allyl phthalimides (4) our results are the best reported to date on these types of substrates (Figure 7).
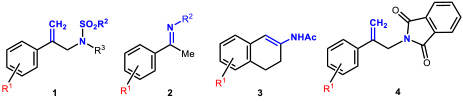 Figure 7. Substrates hydrogenated with Iridium-MaxPHOX complexes with very high enantioselectivity (up to 99% ee).
Figure 7. Substrates hydrogenated with Iridium-MaxPHOX complexes with very high enantioselectivity (up to 99% ee).
In the case of N-alkyl imines a novel C-metalleted was prepared affording excellent enantiomeric excesses (Figure 8). International recognition of our work in the development of P-stereogenic ligands was the publication of a review in the prestigious journal Accounts of Chemical Research of the American Chemical Society. This article addresses our research in the field of asymmetric catalysis during the last decade.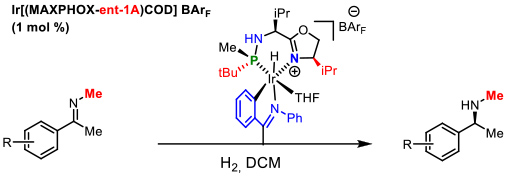 Figure 8. Catalytic asymmetric hydrogenation of N-methyl imines. E. Salomo, A. Gallen, G. Sciortino, G. Ujaque, A. Grabulosa, A. Lledos, A. Riera and X. Verdaguer, J. Am. Chem. Soc. 2018, 140, 16967-16970
Figure 8. Catalytic asymmetric hydrogenation of N-methyl imines. E. Salomo, A. Gallen, G. Sciortino, G. Ujaque, A. Grabulosa, A. Lledos, A. Riera and X. Verdaguer, J. Am. Chem. Soc. 2018, 140, 16967-16970
2. Synthesis of bioactive compounds.
The second main area of research of the group is the synthesis of bioactive compounds. On one side, we have applied the methodologies described above to the synthesis of known drugs. On the other, we have co-led collaborative projects with biology groups in order to test potentially active new compounds.
- 2.1. Application of our methodologies to drug synthesis. The synthetic methodologies developed in our group have allowed the formal synthesis of several enantiomerically enriched chiral pharmaceutically active compounds such as Rotigotine, Sarkomycin, Lorcaserin, OTS514 and LY-404187 (Figure 9).
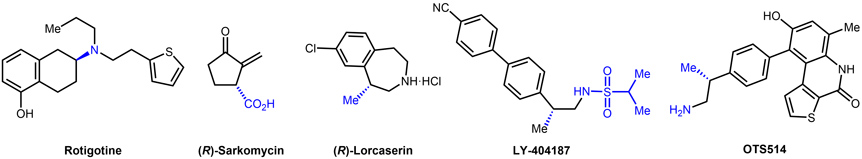 Figure 9. Know drugs synthetized using the asymmetric hydrogenation methodology developed in the group.
Figure 9. Know drugs synthetized using the asymmetric hydrogenation methodology developed in the group.
- 2.2. Hormone analogues. Back in 2011, we started a project in collaboration with BCN Peptides and Maria Macias (IRB Barcelona). The aim of the project was the synthesis of new peptide hormones with improved selectivity and stability. Somatostatin (SRIF) and Cortistatin (CST) are important peptide hormones implicated in multiple biological functions. Its broad anti-secretory profile has led SRIF-14 to clinical practice for the treatment of hypersecretory tumors and gastrointestinal disorders. CST binds to the same receptors (no specific receptor has been found) but shows distinctive anti-inflammatory and immuno-regulatory properties than SRIF. However, the potential therapeutic application of the natural hormones cannot be exploited in full, due to their short half-life in plasma.
Our approach was to introduce site directed modifications into the natural sequence. We found that by fine-tuning the electronic and steric properties of specific aromatic residues (X1-X3), we could obtain peptides with high receptor selectivities and restricted conformations. Using unnatural amino acid such as L-mesityl alanine or L-3-(3’,5’-difluorophenyl)-alanine (Dfp) in positions X1-3 we developed new somatostatin analogues with improved stability and selectivity towards SSTR2.
Recently, we have synthesized new CST analogues with excellent immunoregulatory and anti-CD (Crohn's disease) activity and longer half-life than the native hormones. We have performed a conformational study of the new compounds by NMR and found that the immunoregulatory activity can be related with the CST 3D structure in solution, accounting for its differential pharmacological profile with respect to SRIF. One of these peptides (Figure 10) have potential applications in the treatment of inflammatory diseases such as Crohn's disease.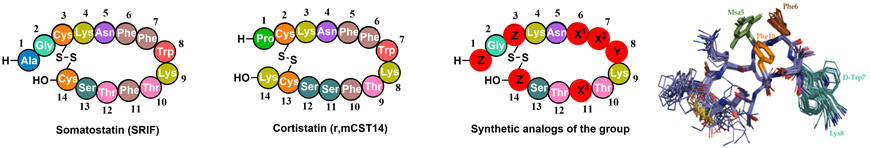 Figure 10. Amino acid sequence of Somatostatin (SRIF) and Cortistatin (CST). General structure of our synthetic analogs. Superposition of the minor energy structures of our anti-inflammatory peptide according to the NMR experimental data. (Rol, A.; Todorovski, T.; Martin-Malpartida, P.; Escola, A.; Gonzalez-Rey, E.; Aragon, E.; Verdaguer, X.; Valles-Miret, M.; Farrera-Sinfreu, J.; Puig, E.; Fernandez-Carneado, J.; Ponsati, B.; Delgado, M.; Riera, A.; Macias, M. J., Nat. Commun. 2021, 12, 1869).
Figure 10. Amino acid sequence of Somatostatin (SRIF) and Cortistatin (CST). General structure of our synthetic analogs. Superposition of the minor energy structures of our anti-inflammatory peptide according to the NMR experimental data. (Rol, A.; Todorovski, T.; Martin-Malpartida, P.; Escola, A.; Gonzalez-Rey, E.; Aragon, E.; Verdaguer, X.; Valles-Miret, M.; Farrera-Sinfreu, J.; Puig, E.; Fernandez-Carneado, J.; Ponsati, B.; Delgado, M.; Riera, A.; Macias, M. J., Nat. Commun. 2021, 12, 1869).
- 2.3 Synthesis of protein inhibitors. The collaboration with Eduard Batlle (IRB Barcelona) also started many years ago, providing his group the TGF-β inhibitors necessary for their research. This collaboration was extremely fruitful, leading to important papers related to the effect of TGF-β inhibition in the treatment of colon cancer metastasis. In this context, we realized that the low water solubility of these compounds was hampering their use. Therefore, we developed new TGF-β inhibitors to be used as soluble pro-drug. Serendipitously, we found that the precursor of these pro-drugs, which we named HOLY, greatly improved the performance of the immunological treatment of metastatic colon cancer. These results are described in our patent (WO2020104648A2) and will be published in the near future (Figure 11).
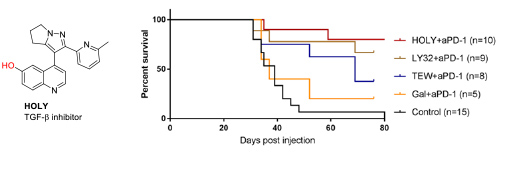 Figure 11. Structure of the TFG-β inhibitor developed by the group. In vivo combination therapy assays of several TGF-β inhibitors with PD1/PDL1 checkpoint immunotherapy
Figure 11. Structure of the TFG-β inhibitor developed by the group. In vivo combination therapy assays of several TGF-β inhibitors with PD1/PDL1 checkpoint immunotherapy
- 2.4 New tools for biology research. PROTACs and protein degraders. An estimated 50-60 % of the genome is considered “undruggable” due to the difficulty to modulate its function using small molecules. During the last decade, the discovery of chimeric drugs able to decrease the levels of a target protein via ubiquitination has changed the field. Proteolysis-targeting chimeras, referred to as PROTACs, are bifunctional compounds that hijack ubiquitinating enzymes to induce the degradation of a target protein by the ubiquitin proteasome pathway. Structurally, PROTACs have one end that binds to the target protein whereas the other is covalently linked to an E3 ubiquitin ligase ligand. We started our project on PROTAC development several years ago, in collaboration with Angel Nebreda (IRB Barcelona). Our work allowed us to identify several PROTACs such as NR-7h (Figure 12) that induce the degradation of both p38α and p38β without affecting other MAPKs, when administered at nanomolar concentrations to the culture medium of several cell lines. In this work we described the optimal linker length for these small molecule PROTACs. We signed a technology transfer agreement with the company Tocris which commercialise protac NT-7h.
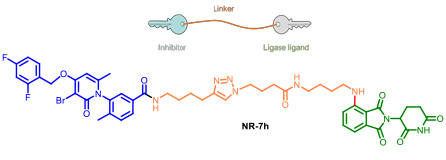
Figure 12. Protac’s general structure. The p38’s protac developed in our group. (Donoghue, C.; Cubillos-Rojas, M.; Gutierrez-Prat, N.; Sanchez-Zarzalejo, C.; Verdaguer, X.; Riera, A.; Nebreda, A. R. Eur. J. Med. Chem. 2020, 201, 112451).
- 2.5 New tools for biology research. Bioorthogonal reactions. Biorthogonal reactions based on click chemistry are having a growing influence both in research and in therapy. Out of the handful of biorthogonal reactions reported to date, the inverse Electron-Demand Diels-Alder (i-EDDA) cycloadditions using 1,2,4,5-tetrazines have been widely applied, becoming extremely useful tools in chemical biology. In this field, we have developed a new reagent (independently and simultaneously with K. Gademan’s group) namely 3-bromo-1,2,4,5-tetrazine. This bromotetrazine proved to be an excellent reagent for the introduction of the tetrazine fragment into biomolecules using nucleophiles such as lysine. We have also described the preparation of several new unnatural amino acids bearing alkynyl- and alkyl-1,2,4,5-tetrazine fragments using a Sonogashira reaction.
To highlight the potential of 3-bromo-1,2,4,5-tetrazine for chemoselective protein labeling and subsequent click-to-release (CtR) drug liberation, we selected the monoclonal antibody Trastuzumab, a frontline in HER2+ breast cancer treatment. The resulting labeled residues reacted with strained dienophiles such as TCO-Doxorubicin (trans-cyclooctene-2-yl doxorubicinyl carbamate) triggering the release of the drug (Figure 13).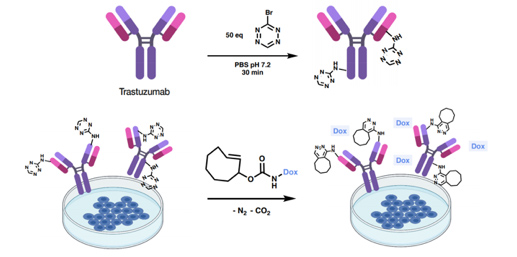
Figure 13. Bioorthogonal liberation of doxorubicin and subsequent cytotoxicity. Labeling of Trastuzumab using 50 equivalents of bromotetrazine and release of doxorubicin (red star) by the reaction of Trastuzumab-1 and TCO-Dox in BT474 (HER2+) cell culture. (Ros, E.; Bellido, M.; Verdaguer, X.; Ribas de Pouplana, L.; Riera, A. Bioconjugate Chem. 2020, 31, 93
Selected publications
Projects
"Enfoques integradores en síntesis química: nuevas estrategias catalíticas y síntesis de compuestos bioactivos para modificaciones específicas de proteínas”, financiado por el Ministerio de Ciencia, Innovación y Universidades mediante la convocatoria 2023 de «PROYECTOS DE GENERACIÓN DE CONOCIMIENTO». Referencia: PID2023-152296OB-

“Desarrollo de nueva metodología sintética y aplicación a la síntesis de compuestos con actividad biológica” (SINESBIO), cofinanciado por el Ministerio de Ciencia, Innovación y Universidades- Agencia Estatal de Investigacióny por el Fondo Europeo de Desarrollo Regional (FEDER) de la Unión Europea. Referencia: CTQ2017-87840-P.
"Synthesis of biologically active compounds. New synthetic methodology, hormone analogs and directed drug release" (SINBIOAC), cofinanciado por el Ministerio de Economía y Competitividad y por el Fondo Europeo de Desarrollo Regional (FEDER) de la Unión Europea. Referencia: CTQ2014-56361-P (MINECO/FEDER, UE).
Suport obtingut de l’FSE a través de les Ajuts per a la contractació de personal investigador novell (FI). El Fons Social Europeu dona suport a la creació d’ocupació, ajuda a las persones a aconseguir millors llocs de treball i garanteix oportunitats laborals més justes per a la ciutadania de la Unió Europea.
“Nuevas herramientas para catálisis asimétrica y química biológica” (TACCBIO), financiado por la Agencia Estatal de Investigación (PID2020-115074GB-I00/ AEI /10.13039/501100011033).
“Un nuevo inhibidor del receptor tipo 1 (ALK5) del TGF-beta; para el tratamiento del cáncer metastásico y la fibrosis”, cofinanciado por el Ministerio de Ciencia e Innovación - Agencia Estatal de Investigación (AEI) y por la Unión Europea "NextGenerationEU" mediante el Plan de Recuperación, Transformación y Resiliencia (PRTR). PDC2021-121226-I00/MCIN/AEI/10.13039/501100011033/NextGenerationEU/PRTR

Grup de Recerca Consolidat "Modelització Molecular i Bioinformàtica" (SGR-Cat 2021) de l’Agència de Gestió d'Ajuts Universitaris i de Recerca (AGAUR). Referencia 2021 SGR 00866