Images
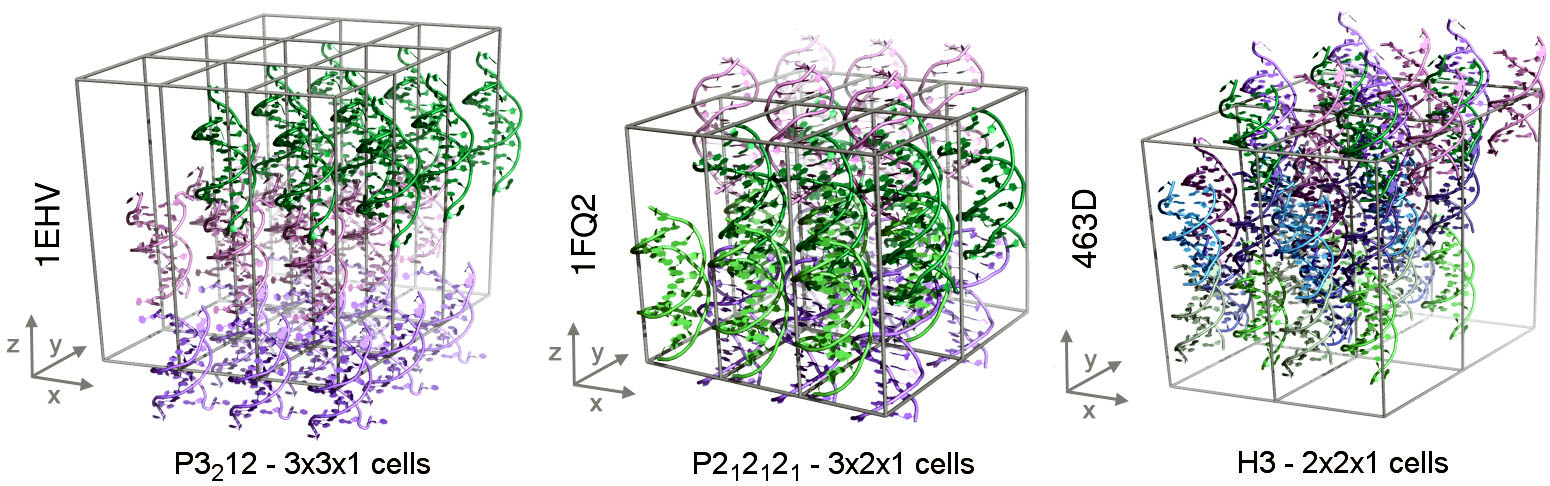
L'avanç aconseguit per investigadors de l'IRB Barcelona permet estudiar la funció de cada component molecular en l'estabilitat i conformació dels vidres d'ADN.
Les noves tècniques de simulacions moleculars reduirien el temps i el cost d'obtenció de cristalls en laboratori.
La cristal·lografia de raigs X ha estat la tècnica més usada des del naixement de la biologia estructural per determinar l'estructura en tres dimensions de les biomolècules, els compostos químics que es troben en els organismes vius. Aquesta tècnica podria optimitzar-se si s’aconeguessin conèixer les interaccions entre les biomolècules amb el seu entorn cristal·lí i les forces moleculars que estabilitzen els vidres.
Un estudi publicat a la revista Chem, del grup Cell, i elaborat per investigadors de l'Institut de Recerca Biomèdica (IRB Barcelona), ha aconseguit per primer cop simulacions estables de cristalls d'ADN. Aquesta fita ha permès explicar la importància dels additius químics que s'usen experimentalment per aconseguir les condicions de cristal·lització i obtenir cristalls estables en el laboratori.
"El primer beneficiari d'estudi és la comunitat de biofísics i fisicoquímics computacionals, que compten ara amb una referència i protocols clars per obtenir simulacions estables de cristalls d'ADN", afirma Pablo D. Dans, investigador postdoctoral de l'IRB Barcelona.
L'estudi liderat per Modesto Orozco, cap del laboratori de Modelització Molecular i Bioinformàtica, fa la descripció més detallada a nivell atòmic de les propietats de sistemes cristal·lins amb ADN presentada fins la data.
"A llarg termini, la simulació de diversos cristalls obtinguts en diferents condicions experimentals hauria de permetre anticipar i predir l'efecte d'un additiu químic donat, i guiar als cristal·lògrafs en els seus experiments reduint considerablement els costos i els temps d'obtenció de cristalls", subratlla Modesto Orozco, catedràtic de la Facultat de Química de la Universitat de Barcelona, que lidera un dels laboratoris referents al món en computació i simulació de biomolècules.
L'estudi ha rebut fons del Consell Europeu de Recerca (ERC en les seves sigles en anglès), a través del projecte ERC Advanced Grant (SimDNA) atorgat a Modesto Orozco, i del Consorci Europeu de Centres de Supercomputació a través de la beca PRACE 12th Call, atorgada als tres autors de l'estudi.
Article de referència:
Antonija Kuzmanic, Pablo D. Dans and Modesto Orozco.
An in-depth look at DNA crystals through the prism of molecular dynamics simulations
Chem (2019) DOI: 10.1016/j.chempr.2018.12.007
IRB Barcelona
L’Institut de Recerca Biomèdica (IRB Barcelona) treballa per aconseguir una vida lliure de malalties. Desenvolupa una recerca multidisciplinària d’excel·lència per curar el càncer i altres malalties vinculades a l'envelliment. Treballa establint col·laboracions amb la indústria farmacèutica i els principals hospitals per fer arribar els resultats de la recerca a la societat a través de la transferència de tecnologia, i du a terme diferents iniciatives de divulgació científica per mantenir un diàleg obert amb la ciutadania. L’IRB Barcelona és un centre internacional que acull al voltant de 400 investigadors de més de 30 nacionalitats. Reconegut com a Centre d'Excel·lència Severo Ochoa des de 2011, és un centre CERCA i membre del Barcelona Institute of Science and Technology (BIST).