Images

Kennedy’s disease leads to progressive muscle wastage. It is a condition that affects only men and it appears between 40 and 50 years of age. There is no specific treatment available.
In individuals with this disease, muscle cells and motor neurons die over the years because they accumulate a protein that is mutated.
Researchers at IRB Barcelona discover that this protein has a self-protective mechanism through which the deleterious effects of the mutation are delayed.
Knowledge of the minute details of the proteins that are linked to diseases is crucial if we are to discover therapeutic targets and thus pave the way for possible treatments. Such knowledge gains even more relevance when dealing with rare diseases that have received little attention and for which no treatments are available, such as the case of Kennedy’s disease. This week Xavier Salvatella, ICREA researcher at the Institute for Research in Biomedicine (IRB Barcelona), in collaboration with scientists from the University of Florence (Italy), has described a molecular system of protection that involves the androgen receptor protein, a molecule that is mutated in patients with Kennedy’s disease and which cause progressive muscle wastage.
The finding brings in-depth molecular insights that can lead to new studies and bring researchers closer to finding a therapeutic target for Kennedy’s disease. The study has been published in Biophysical Journal, part of the Cell group.
Androgen Receptor Structure Animation (Bursa Topal)
In Kennedy’s disease, the muscle cells and motor neurons—the latter linked to muscle function too—are damaged as a result of the accumulation of androgen receptor fibers—a process that causes them to die. "Many aspects of diseases involving aggregates, such as Alzheimer’s and Parkinson’s, are unknown. In this regard, Kennedy’s disease is in a worse position because it is a rare condition," explains Xavier Salvatella, head of the Molecular Biophysics Lab at IRB Barcelona.
The onset of this genetically inherited disease occurs in late adulthood, affecting one in every 40,000 men and causing progressive deterioration of all muscles. Although not fatal, the condition is debilitating, and 20% of those affected eventually need a wheel chair.
Backed by funding from the “Fundación La Marató de TV3” and a Consolidator Grant from the European Research Council, Salvatella studies the sequence of molecular events that causes the aggregation of androgen receptors. He and his team seek to determine the regions of the protein sequence responsible for aggregation in order to identify valid targets to direct therapies and to prevent this process.
Attacking the flanks of the protein
The mutation carried by those affected by Kennedy’s disease causes a repeated and excessively long chain of a specific amino acid, namely glutamine, which impairs the activity of the androgen receptor. This protein, which activates the hormone testosterone, is responsible for triggering the genetic programme that favours the differential characteristics of men: more hair, deep voice, larger hands, etc.
During adolescence, and depending on the extent to which they are affected, boys with the mutation do not fully develop the male phenotype, but this is often not diagnosed. In a second stage, in adulthood, muscle degeneration begins. "It is known that the longer the polyglutamine chain, the earlier the onset of muscle atrophy. What we don’t know and what this study helps us to understand is why the harmful effect is triggered when the length of the poly glutamine exceed 38 residues".
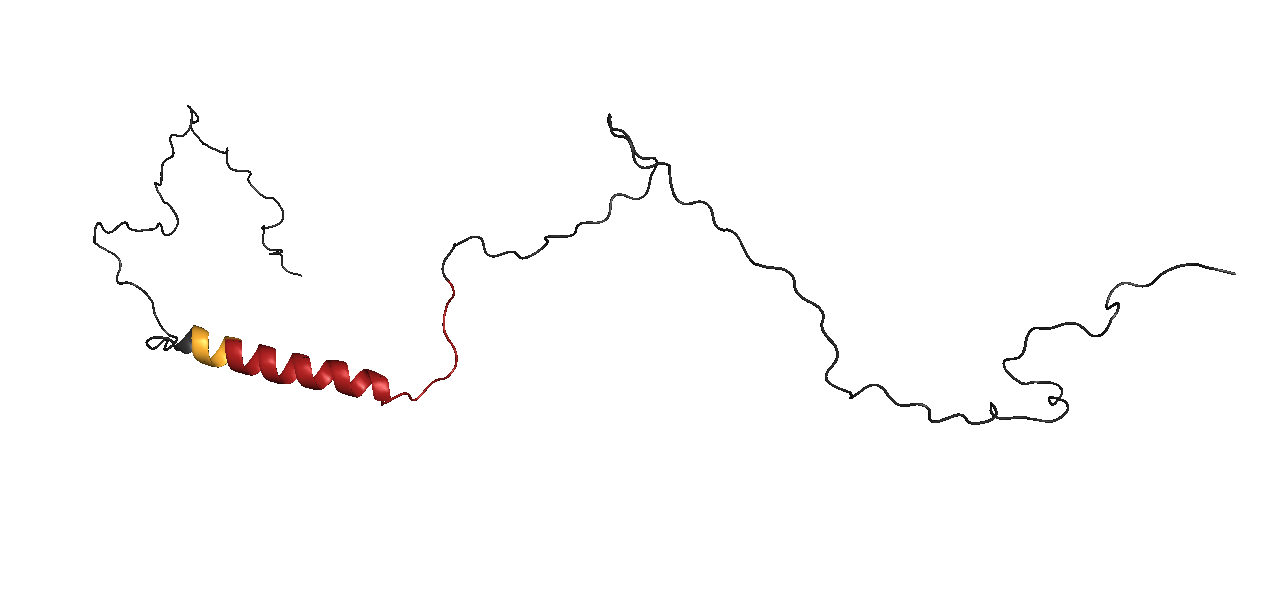
Thanks to access to one of the main Nuclear Magnetic Resonance facilities in Europe, located at the University of Florence, for the first time the scientists have studied the protein in a test tube. They have observed that right next to the glutamine chain there is a region comprised by four leucine residues that allay the effects of the mutation.
The leucine molecules favour the folding of the polyglutamine chain into a helix, a structure that prevents the chains from adhering to one another. However, the impact of the leucine molecule on the glutamine region is limited, and if there are many glutamine amino acids, the chains do not fold. Instead, they stretch out like rods, stick to each other, and end up forming a fibrous wall.
"We have seen that four leucine molecules delay this process. What would happen in the presence of six?" asks Salvatella. "Conceptually speaking, one clever way of delaying the aggregation could be to use drugs to strengthen the effect of the leucine residues that have so much influence on the mutation site that causes the protein to aggregate," reflects Salvatella.
Salvatella’s work indicates that in-depth knowledge of the different protein sequences and how they affect each other may reveal new therapeutic targets. The IRB Barcelona scientist is engaged in several research lines devoted to Kennedy’s disease. His studies involve the development of techniques and studies with mice and with chaperones—proteins that bind to any protein that is about to aggregate. "This is the first article in a series in which we can slowly explain how we think Kennedy’s disease develops and propose therapeutic solutions," he explains.
The Iranian researcher Bahareh Eftekharzadeh is the first author of the paper. She did her PhD at IRB Barcelona funded by an International “la Caixa” Fellowship and is currently doing postdoc work at the University of Harvard. Salvatella’s lab also receives funding from the Ministry of the Economy and Competitiveness, complemented by support from the ERDF and the Government of Catalonia.
Reference article:
Sequence Context influences the Structure and Aggregation Behavior of a PolyQ Tract
B. Eftekharzadeh, A. Piai, G. Chiesa, D. Mungianu, J. García, R. Pierattelli, E. C. Felli, and X. Salvatella
Biophysical Journal (7 June 2016) doi: 10.1016/j.bpj.2016.04.022
About IRB Barcelona
The Institute for Research in Biomedicine (IRB Barcelona) pursues a society free of disease. To this end, it conducts multidisciplinary research of excellence to cure cancer and other diseases linked to ageing. It establishes technology transfer agreements with the pharmaceutical industry and major hospitals to bring research results closer to society, and organises a range of science outreach activities to engage the public in an open dialogue. IRB Barcelona is an international centre that hosts 400 researchers and more than 30 nationalities. Recognised as a Severo Ochoa Centre of Excellence since 2011, IRB Barcelona is a CERCA centre and member of the Barcelona Institute of Science and Technology (BIST).