
Dual appointment with BSC
Research information
Background
Our research interests are focused around the use of comparative genomics and phylogenomics to study the origin, evolution and function of complex biological systems. This includes understanding how specific biochemical pathways, protein complexes or cellular organelles emerged and evolved as well as using this evolutionary information to gain insight into their function.
Through collaborations with experimental groups we apply comparative genomics to discover new mechanisms and genes involved in interesting processes, especially those of clinical relevance (see lines of research). On the technical side, our work often involves the development of new bioinformatics tools and algorithms that we make available to the community.
Research lines
http://cgenomics.org/research/
Phylogenomics and genome evolution
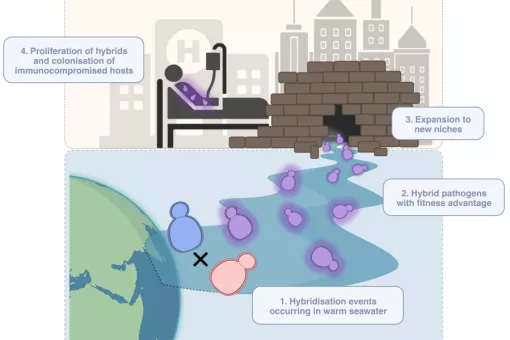
In the genomic era it has been possible to move from the evolutionary analysis of single protein families (phylogenetics) to that of complete genomes and proteomes (phylogenomics). To achieve this transition new tools have been developed that allow the large-scale reconstruction of thousands of phylogenetic trees in an automatic way. Interpreting such type of complex data poses many difficulties and does require the development of novel algorithms, tools, forms of representing the data and even new semantics and concepts. We combine the development of original algorithms to treat phylogenomic data with its application to gain knowledge on problems of biological relevance. We have recently started an ERC-funded project entitled RETVOLUTION: “processes and patterns of reticulate evolution in eukaryotes”, which will combine innovative computational and experimental approaches to infer patterns of reticulate evolution across the eukaryotic tree, and relate this to current biological knowledge. A particular focus is placed on elucidating the role of hybridization in the origin of whole genome duplications, and in facilitating the spread of horizontally transferred genes.
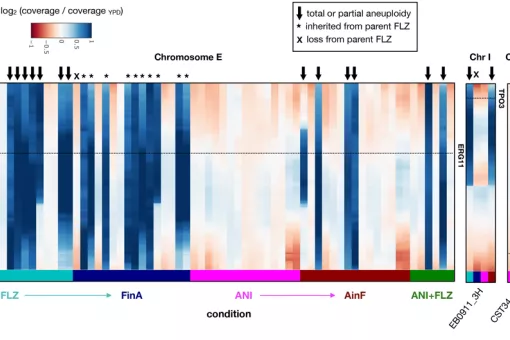
Comparative genomics and population genomics of fungal pathogens
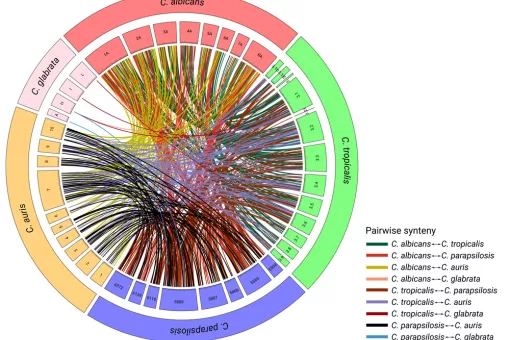
Fungal infections constitute an ever-growing and significant medical problem. Diseases caused by such pathogens range from simple toe nail infections, to life-threatening systemic mycoses in patients with impaired immune systems. The molecular mechanisms driving invasion of mammalian hosts by fungal pathogens poses many scientifically challenging problems, which are as yet little understood. The ability to infect humans has emerged in several lineages throughout the fungal tree of life. Therefore, the problem of elucidating the mechanism for pathogenesis of fungi, as proposed here, can be approached with an evolutionary perspective by detecting specific adaptations in pathogenic lineages. During the last years we have clarified the evolutionary paths to virulence of major fungal pathogens such as Candida glabrata and Candida parapsilosis.
Microbiome-host interactions in health and disease
As a natural progression of our interest in the role of fungi in health and disease, the group started working on the analysis of the microbiome. Our main focus within this field is quantifying and understanding the role of the fungal component, which is usually neglected by mainstream analyses. In the last three years we have engaged in several massive projects, which include the oral microbiome project “Saca La Lengua” (www.sacalalengua.org), with over 1500 samples, the lung microbiome in patients with chronic respiratory syndrome, the lung microbiome in the intensive care unit patient (with artificial ventilators), and the intestinal microbiome in colon cancer. These projects involve analysis of bacterial metagenomes and of the fungal component. For the analysis of the fungal microbiome we have developed novel approaches that overcome existing limitations of the traditional ITS-based sequencing.
Evolutionary genomics of long, non-coding RNAs
Recent genomics analyses have facilitated the discovery of a novel major class of stable transcripts, now called long non-coding RNAs (lncRNAs). A growing number of analyses have implicated lncRNAs in the regulation of gene expression, dosage compensation and imprinting, and there is increasing evidence suggesting the involvement of lncRNAs in various diseases such as cancer. Despite recent advances, however, the role of the large majority of lncRNAs remains unknown and there is current debate on what fraction of lncRNAs may just represent transcriptional noise. Our group has recently embarked into a project, funded by the European Research Council that aims to combine state-of-the-art computational and sequencing techniques in order to elucidate what evolutionary mechanisms are shaping this enigmatic component of eukaryotic genomes.
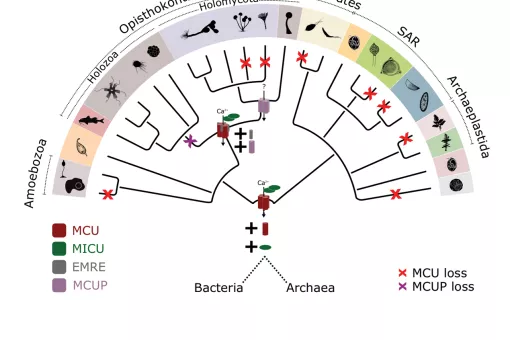
Evolution of eukaryotes
Every eukaryotic organism shows a high level of sub-cellular compartmentalization that is significantly more intricate than the most complex prokaryotic cell. How such degree of complexity came to be is still not fully understood. In this context, endo-symbiotic events with bacterial organisms have been proposed to be the source of a number of organelles including mitochondria, chloroplasts and peroxisomes. Only recently, it has been possible to contrast these hypotheses with the growing availability of completely sequenced genomes and organellar proteomic data. In the last years, we have used large-scale evolutionary analyses to investigate the origin and evolution two most widespread organelles for which an endosymbiotic origin has been proposed: mitochondria and peroxisomes.
Selected publications
Projects
“Reticulate evolution: patterns and impacts of non-vertical inheritance in eukaryotic genomes" (RETVOLUTION), financiado por el European Research Council (ERC) mediante el Programa de Investigación e Innovación de la Unión Europea Horizonte 2020. Referencia: 724173
"Evolución genómica y fenotípica en patógenos del género Candida" (genomePath), cofinanciado por el Ministerio de Ciencia, Innovación y Universidades- Agencia Estatal de Investigación y por el Fondo Europeo de Desarrollo Regional (FEDER) de la Unión Europea. Referencia: PGC2018-099921-B-I00
Grup de Recerca consolidat (SGR 2017-2019) de la Secretaria d'Universitats i Recerca del Departament d'Empresa i Coneixement de la Generalitat de Catalunya. Agencia de Gestió d'Ajuts Universitaris i de Recerca (AGAUR). Referencia: 2017 SGR 423
“Prevenció del càncer colorectal en la població de risc mitjà mitjançant biomarcadors genòmics i microbiòmics (CRIPREV)” financiado por la Convocatòria 2017 del Pla (PERIS) ‐ Acció Instrumental de Programes de Recerca Orientats Estratègic de Recerca i Innovació en Salut - Generalitat de Catalunya. Referencia: STL002/16/00398
“The flagellate Amastigomonas sp. MONTSE19P8 from Montserrat, a key species in eukaryotic evolution”, financiado por el Institut d’Estudis Catalans en la convocatoria “Genome assemblies for eukaryotic species within the Catalan territories”, 2020.
“Origen, evolución y adaptación en patógenos del género Candida: una aproximación genomica y transcriptómica”, proyecto PID2021-126067NB-I00 financiado por MCIN/ AEI /10.13039/501100011033/ y por FEDER Una manera de hacer Europa.
“LactAs: desarrollo de un bioterapéutico vivo recombinante para el tratamiento del asma grave”, proyecto CPP2021-008552 financiado por MCIN/ AEI /10.13039/501100011033/ por la Unión Europea NextGenerationEU/ PRTR
“Nuevos compuestos antifúngicos contra Candida multiresistente”, proyecto PDC2022-133266-I00 financiado por MCIN/ AEI /10.13039/501100011033/ por la Unión Europea NextGenerationEU/ PRTR
“Disrupting drug resistance by exploiting collateral sensitivity to design novel”, proyecto PCI2022-135066-2 financiado por MCIN/ AEI /10.13039/501100011033/ por la Unión Europea NextGenerationEU/ PRTR
“Acquisition of antifungal resistance in pathogenic Candida spp.: evolutionary paths, fitness trade-offs, and interplay with the immune system”. Fundació La Caixa. Referencia: LCF/PR/HR21/52410006
"MICROSCREEN_CRC: Microbial biomarker test for early diagnosis of colorectal cancer (MiCRC)". Fundació La Caixa. Referencia: CI23-20260
"Metagenomic profiling from fecal immunochemical test tubes to identify early players and diagnostic markers in colorectal cancer" funded by Fundación Científica AECC. Reference: PRYGN234923GABA
"Computational multiplexing to optimise next-generation sequencing" funded by the European Union. Reference: 101123165